Molecular Magnetism
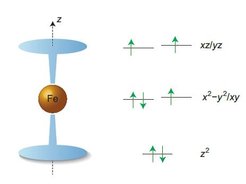
 center with axial symmetry (cooperation with the group of Jeffrey Long, Berkeley, California, Nature. Chem. 5, 577-581, 2013) affords the orbital electronic configuration (right) –the origin of a large magnetic moment along the z-direction (left). Such molecules can find potential use as molecular storage devices (q-bits) or quantum computers.")
Molecular magnetism is a multidisciplinary field that combines synthesis of molecules with desired magnetic properties and theoretical analysis aiming at mechanistic understanding of the relation of these properties with the electronic structure. Spectacular advances in the development and implementation in the program ORCA of spin-dependent relativistic correlated electronic structure methods for larger systems by Prof. Dr. Frank Neese and his developer team supplied the infrastructure and serves as a firm theoretical basis for this project. The combination of experiment and theory on a one-to-one basis allows to both interpret and to predict (rationally design) magnetic properties of target magnetic materials. Molecular magnetism has a big impact on closely related disciplines such as molecular electronics and chemical reactivity (homogeneous and heterogeneous catalysis). The latter has been inspired by the action of transition metal spin centers at the active sites in enzymes such as the spitting of water on a four nuclear mixed-valence manganese complex (Photosystem II) and oxygen and hydrogen atom transfer in heme and non-heme high-valence iron enzymes leading to molecules for energy storage. All this motivated the creation in 2013 of a group on molecular magnetism at the institute. Basic tools in the work on this project have been: i) computational protocols allowing to extract magnetic and spin-Hamiltonian (SH) parameters from ab intio correlated wave functions [1a], and ii) an ab initio based ligand field theory (AILFT) that allows to relate the magnetic parameters with the nature of the metal-ligand bonds and the complex geometries [1b].
Open d- or f-shell transition metal or lanthanides and actinides in complexes give rise to an unique response to an external magnetic field governed by the preferred alignments of their magnetic moments. This property is termed magnetic anisotropy. In axial symmetry this anisotropy is described by one parameter D which quantifies the zero-field splitting of the 2S+1 sublevels M ( -S<M<S ) of the spin S (E=DM2) of the non-relativistic ground state. Magnetic anisotropy arises from coupling between orbital and spin angular momenta. Below a given temperature (called the blocking temperature, TB) the resulting total angular momentum and its magnetization induced by an external magnetic field may persists after switching off the field. The specific life-time of this magnetic state -the relaxation time depends on the magnitude of the orbital angular momentum and its coupling to its dissipative surrounding (thermal bath). Orbital momenta are maximal in atoms and ions with orbitally degenerate ground states, such as FeI 4F(d7) or FeII 2D(d6). However, because of their spherical symmetry, such orbital moments are isotropic. Anisotropic magnetic moments are intrinsic for molecules possessing axial (four of threefold) symmetries, which in addition, are in orbitally degenerate or nearly-denerate ground states. Spin-orbit coupling introduces atomic like orbital moments which couple with the spin and this leads to an entirely anisotropic magnetization (Ising anisotropy). This magnetization is maximal along the axis of quantization (the easy axis, D<0) and (in weak magnetic fields) very small in directions perpendicular to it.
The project focuses on magnetic properties of transition metal complexes with open d-shells. Open d- or f-shell transition metal or a lanthanide complex can give rise to an unique response to an external magnetic field governed by the preferred alignments of their magnetic moments. This property is termed magnetic anisotropy. In axial symmetry this anisotropy is described by one parameter D which quantifies the zero-field splitting of the 2S+1 sublevels M ( -S≤M≤S ) of the spin S (E=DM2) within a given electronic state (normally the non-relativistic ground state). Magnetic anisotropy arises from coupling between orbital and spin angular momenta. Below a given temperature (called the blocking temperature, TB) the resulting total angular momentum and its magnetization induced by an external magnetic field may persist after switching off the field. The specific life-time of this magnetic state -the relaxation time depends on the magnitude of the orbital angular momentum and its coupling to its dissipative surrounding (thermal bath). Orbital momenta are maximal in atoms and ions with orbitally degenerate ground states, such as Co(II) 4F or Fe(II) 2D. However, because of the spherical symmetry, such orbital moments are isotropic and do not create magnetic anisotropies. Anisotropic magnetic moments are intrinsic for molecules possessing axial four of threefold symmetries, which in addition, are in orbitally degenerate ground states. First order spin-orbit coupling in such systems introduces atomic like orbital moments which couple with the spin and this leads to an entirely anisotropic magnetization. This magnetization is maximal along the axis of quantization (the easy axis, D<0) and (in weak magnetic fields) very small in directions perpendicular to it (Ising type anisotropy). A series of pseudo-tetrahedral Fe(II) complexes of this type has been theoretically predicted [1] by first principles calculations and synthesized and magnetically characterized [2,3] at about the same time (see Figure 1). These were shown to display slow relaxation of the magnetization and were termed single ion magnets (SIM). These discoveries opened a new field in magneto chemistry. The ultimate goal of the studies carried out within this project is to increase the blocking temperature and the relaxation time making such systems potential candidates for magnetic memory devices. Systems with magnetically bi stable ground states in molecules well isolated from their dissipative surroundings can also display quantum coherence and are therefore of potential interest in future quantum computers.
Based on the infrastructure given by the development and implementation of multi reference electronic structure methods in the ORCA program by Prof. Frank Neese and his group, we developed and tested computational protocols for first principles computations of the magnetic and spectroscopic properties of SIM which allow to both interpret and predict such phenomena [4]. We applied these tools to the interpretation of existing SIM and to assist rational design of novel ones with improved magnetic properties. We studied trigonal four coordinate (Figure 1, [4]) and pseudo linear two coordinate Fe(II) complexes (Figure 2, [5,6]) the latter displaying magnetic anisotropies on unprecedented size.
A fruitful scientific exchange with our collaborators (Prof. J. Long, Berkeley, USA) culminated in understanding of the magnetic properties of these class of compounds and the first SIM displaying a magnetic hysteresis and magnetic blocking temperature TB=4K (Figure 3) [7].